SIMPAL - SIMple Plasmid ALignemnt
SIMPAL is a small script written in R with the purpose for simple N->1 gene alignment and mutation analysis. The idea is to have a handy tool to analyze some tens to thousands of equivalent data sets containing just one or a few mutation as it is the situation within a gene library derived from random mutagenesis protocols. The correct ORF is determined by a unique identifier (Fig. 1) , a short recognition sequence of 12-15 nucleotides upstream of the target gene which also serves as the starting point for further alignment. Before you can start you need to tell the script just three information. First of course the wild-type sequence within a certain plasmid system (intended) or even as stand-alone gene both written as plain text into a single file. Secondly the unique identifier also given as a plain text within a another file. The third information is the directory where the script could find your sequencing library in FASTA-format. Assuming everything works well, the output of the script contains some general statistics about the absolute number and distribution of single and multiple mutations. Moreover, a so called level plot and its corresponding Nx20 matrix are generated representing a mutation frequency diagram with a amino acid to amino acid assignment. The results from a mutation analysis of up to several thousands of data sets is thus concentrated within a single plot (Fig. 2).
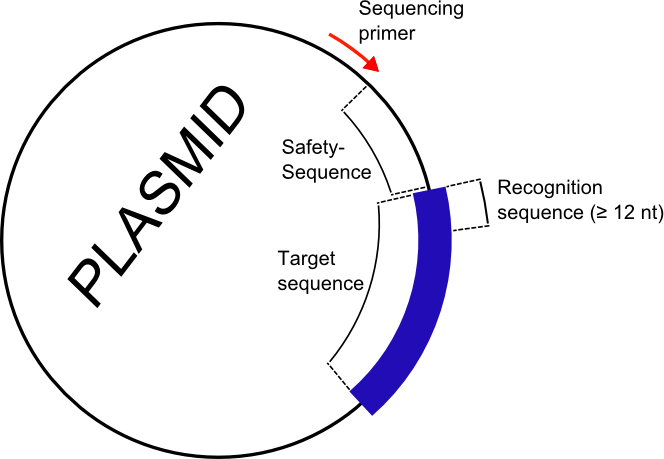
using SIMPAL (Graphic made with Inkscape).
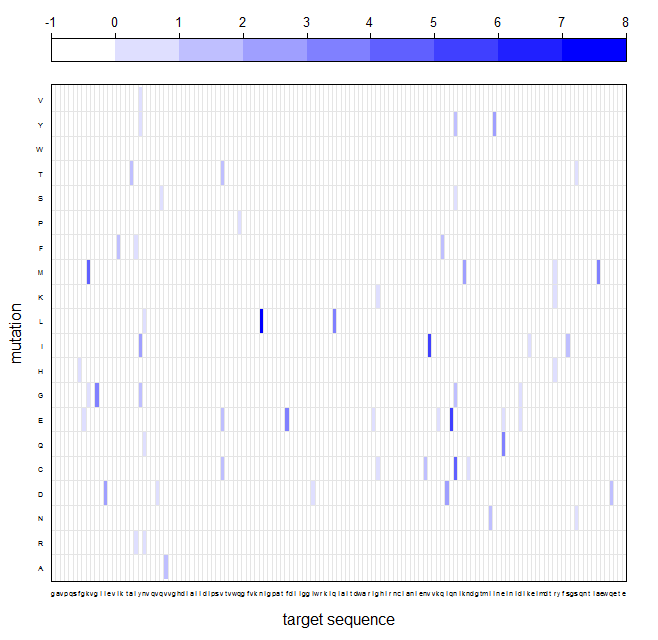
SIMPAL source code